4.3.5. Calculating root mean square quantities — MDAnalysis.analysis.rms
- Author:
Oliver Beckstein, David L. Dotson, John Detlefs
- Year:
2016
- Copyright:
Lesser GNU Public License v2.1+
Added in version 0.7.7.
Changed in version 0.11.0: Added RMSF analysis.
Changed in version 0.16.0: Refactored RMSD to fit AnalysisBase API
The module contains code to analyze root mean square quantities such
as the coordinat root mean square distance (RMSD) or the
per-residue root mean square fluctuations (RMSF).
This module uses the fast QCP algorithm [Theobald2005] to calculate
the root mean square distance (RMSD) between two coordinate sets (as
implemented in
MDAnalysis.lib.qcprot.CalcRMSDRotationalMatrix()).
When using this module in published work please cite [Theobald2005].
See also
MDAnalysis.analysis.alignaligning structures based on RMSD
MDAnalysis.lib.qcprotimplements the fast RMSD algorithm.
4.3.5.1. Example applications
4.3.5.1.1. Calculating RMSD for multiple domains
In this example we will globally fit a protein to a reference structure and investigate the relative movements of domains by computing the RMSD of the domains to the reference. The example is a DIMS trajectory of adenylate kinase, which samples a large closed-to-open transition. The protein consists of the CORE, LID, and NMP domain.
superimpose on the closed structure (frame 0 of the trajectory), using backbone atoms
calculate the backbone RMSD and RMSD for CORE, LID, NMP (backbone atoms)
The trajectory is included with the test data files. The data in
RMSD.results.rmsd is plotted with matplotlib.pyplot.plot() (see Figure RMSD plot figure):
import MDAnalysis
from MDAnalysis.tests.datafiles import PSF,DCD,CRD
u = MDAnalysis.Universe(PSF,DCD)
ref = MDAnalysis.Universe(PSF,DCD) # reference closed AdK (1AKE) (with the default ref_frame=0)
#ref = MDAnalysis.Universe(PSF,CRD) # reference open AdK (4AKE)
import MDAnalysis.analysis.rms
R = MDAnalysis.analysis.rms.RMSD(u, ref,
select="backbone", # superimpose on whole backbone of the whole protein
groupselections=["backbone and (resid 1-29 or resid 60-121 or resid 160-214)", # CORE
"backbone and resid 122-159", # LID
"backbone and resid 30-59"]) # NMP
R.run()
import matplotlib.pyplot as plt
rmsd = R.results.rmsd.T # transpose makes it easier for plotting
time = rmsd[1]
fig = plt.figure(figsize=(4,4))
ax = fig.add_subplot(111)
ax.plot(time, rmsd[2], 'k-', label="all")
ax.plot(time, rmsd[3], 'k--', label="CORE")
ax.plot(time, rmsd[4], 'r--', label="LID")
ax.plot(time, rmsd[5], 'b--', label="NMP")
ax.legend(loc="best")
ax.set_xlabel("time (ps)")
ax.set_ylabel(r"RMSD ($\AA$)")
fig.savefig("rmsd_all_CORE_LID_NMP_ref1AKE.pdf")
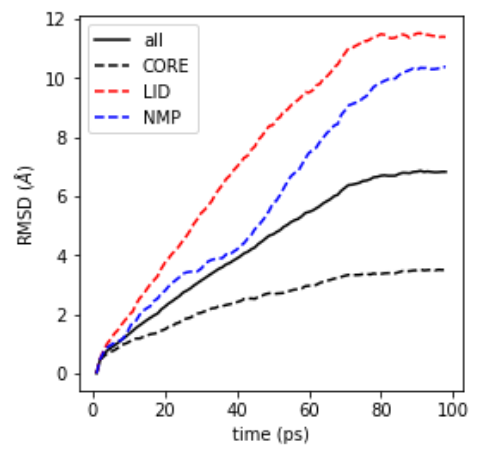
RMSD plot for backbone and CORE, LID, NMP domain of the protein.
4.3.5.2. Functions
- MDAnalysis.analysis.rms.rmsd(a, b, weights=None, center=False, superposition=False)[source]
Returns RMSD between two coordinate sets a and b.
a and b are arrays of the coordinates of N atoms of shape \(N times 3\) as generated by, e.g.,
MDAnalysis.core.groups.AtomGroup.positions().Note
If you use trajectory data from simulations performed under periodic boundary conditions then you must make your molecules whole before performing RMSD calculations so that the centers of mass of the mobile and reference structure are properly superimposed.
- Parameters:
a (array_like) – coordinates to align to b
b (array_like) – coordinates to align to (same shape as a)
weights (array_like (optional)) – 1D array with weights, use to compute weighted average
center (bool (optional)) – subtract center of geometry before calculation. With weights given compute weighted average as center.
superposition (bool (optional)) – perform a rotational and translational superposition with the fast QCP algorithm [Theobald2005] before calculating the RMSD; implies
center=True.
- Returns:
rmsd – RMSD between a and b
- Return type:
Notes
The RMSD \(\rho(t)\) as a function of time is calculated as
\[\rho(t) = \sqrt{\frac{1}{N} \sum_{i=1}^N w_i \left(\mathbf{x}_i(t) - \mathbf{x}_i^{\text{ref}}\right)^2}\]It is the Euclidean distance in configuration space of the current configuration (possibly after optimal translation and rotation) from a reference configuration divided by \(1/\sqrt{N}\) where \(N\) is the number of coordinates.
The weights \(w_i\) are calculated from the input weights weights \(w'_i\) as relative to the mean:
\[w_i = \frac{w'_i}{\langle w' \rangle}\]Example
>>> import MDAnalysis as mda >>> from MDAnalysis.analysis.rms import rmsd >>> from MDAnalysis.tests.datafiles import PSF, DCD >>> u = mda.Universe(PSF, DCD) >>> bb = u.select_atoms('backbone') >>> A = bb.positions.copy() # coordinates of first frame >>> _ = u.trajectory[-1] # forward to last frame >>> B = bb.positions.copy() # coordinates of last frame >>> rmsd(A, B, center=True) 6.838544558398293
Changed in version 0.8.1: center keyword added
Changed in version 0.14.0: superposition keyword added
4.3.5.3. Analysis classes
- class MDAnalysis.analysis.rms.RMSD(atomgroup, reference=None, select='all', groupselections=None, weights=None, weights_groupselections=False, tol_mass=0.1, ref_frame=0, **kwargs)[source]
Class to perform RMSD analysis on a trajectory.
The RMSD will be computed for two groups of atoms and all frames in the trajectory belonging to atomgroup. The groups of atoms are obtained by applying the selection selection select to the changing atomgroup and the fixed reference.
Note
If you use trajectory data from simulations performed under periodic boundary conditions then you must make your molecules whole before performing RMSD calculations so that the centers of mass of the selected and reference structure are properly superimposed.
Run the analysis with
RMSD.run(), which stores the results in the arrayRMSD.results.rmsd.Changed in version 1.0.0:
save()method was removed, usenp.savetxt()onRMSD.results.rmsdinstead.Changed in version 2.0.0:
rmsdresults are now stored in aMDAnalysis.analysis.base.Resultsinstance.Changed in version 2.8.0: introduced
get_supported_backends()allowing for parallel execution onmultiprocessinganddaskbackends.- Parameters:
atomgroup (AtomGroup or Universe) – Group of atoms for which the RMSD is calculated. If a trajectory is associated with the atoms then the computation iterates over the trajectory.
reference (AtomGroup or Universe (optional)) – Group of reference atoms; if
Nonethen the current frame of atomgroup is used.select (str or dict or tuple (optional)) –
The selection to operate on; can be one of:
any valid selection string for
select_atoms()that produces identical selections in atomgroup and reference; ora dictionary
{'mobile': sel1, 'reference': sel2}where sel1 and sel2 are valid selection strings that are applied to atomgroup and reference respectively (theMDAnalysis.analysis.align.fasta2select()function returns such a dictionary based on a ClustalW or STAMP sequence alignment); ora tuple
(sel1, sel2)
When using 2. or 3. with sel1 and sel2 then these selection strings are applied to atomgroup and reference respectively and should generate groups of equivalent atoms. sel1 and sel2 can each also be a list of selection strings to generate a
AtomGroupwith defined atom order as described under Ordered selections).groupselections (list (optional)) –
A list of selections as described for select, with the difference that these selections are always applied to the full universes, i.e.,
atomgroup.universe.select_atoms(sel1)andreference.universe.select_atoms(sel2). Each selection describes additional RMSDs to be computed after the structures have been superimposed according to select. No additional fitting is performed.The output contains one additional column for each selection.Note
Experimental feature. Only limited error checking implemented.
weights ({“mass”,
None} or array_like (optional)) –“mass” will use masses as weights for both select and groupselections.
Nonewill weigh each atom equally for both select and groupselections.
3. If 1D float array of the same length as atomgroup is provided, use each element of the array_like as a weight for the corresponding atom in select, and assumes
Nonefor groupselections.weights_groupselections (False or list of {“mass”,
Noneor array_like} (optional)) –1.
Falsewill apply imposed weights to groupselections fromweightsoption ifweightsis either"mass"orNone. Otherwise will assume a list of length equal to length of groupselections filled withNonevalues.2. A list of {“mass”,
Noneor array_like} with the length of groupselections will apply the weights to groupselections correspondingly.tol_mass (float (optional)) – Reject match if the atomic masses for matched atoms differ by more than tol_mass.
ref_frame (int (optional)) – frame index to select frame from reference
verbose (bool (optional)) – Show detailed progress of the calculation if set to
True; the default isFalse.
- Raises:
SelectionError – If the selections from atomgroup and reference do not match.
TypeError – If weights or weights_groupselections is not of the appropriate type; see also
MDAnalysis.lib.util.get_weights()ValueError – If weights are not compatible with atomgroup (not the same length) or if it is not a 1D array (see
MDAnalysis.lib.util.get_weights()). AValueErroris also raised if the length of weights_groupselections are not compatible with groupselections.
Notes
The root mean square deviation \(\rho(t)\) of a group of \(N\) atoms relative to a reference structure as a function of time is calculated as
\[\rho(t) = \sqrt{\frac{1}{N} \sum_{i=1}^N w_i \left(\mathbf{x}_i(t) - \mathbf{x}_i^{\text{ref}}\right)^2}\]The weights \(w_i\) are calculated from the input weights weights \(w'_i\) as relative to the mean of the input weights:
\[w_i = \frac{w'_i}{\langle w' \rangle}\]The selected coordinates from atomgroup are optimally superimposed (translation and rotation) on the reference coordinates at each time step as to minimize the RMSD. Douglas Theobald’s fast QCP algorithm [Theobald2005] is used for the rotational superposition and to calculate the RMSD (see
MDAnalysis.lib.qcprotfor implementation details).The class runs various checks on the input to ensure that the two atom groups can be compared. This includes a comparison of atom masses (i.e., only the positions of atoms of the same mass will be considered to be correct for comparison). If masses should not be checked, just set tol_mass to a large value such as 1000.
See also
Added in version 0.7.7.
Changed in version 0.8: groupselections added
Changed in version 0.16.0: Flexible weighting scheme with new weights keyword.
Deprecated since version 0.16.0: Instead of
mass_weighted=True(removal in 0.17.0) use newweights='mass'; refactored to fit with AnalysisBase APIChanged in version 0.17.0: removed deprecated mass_weighted keyword; groupselections are not rotationally superimposed any more.
Changed in version 1.0.0: filename keyword was removed.
- results.rmsd
Contains the time series of the RMSD as an N×3
numpy.ndarrayarray with content[[frame, time (ps), RMSD (A)], [...], ...].Added in version 2.0.0.
- rmsd
Alias to the
results.rmsdattribute.Deprecated since version 2.0.0: Will be removed in MDAnalysis 3.0.0. Please use
results.rmsdinstead.
- classmethod get_supported_backends()[source]
Tuple with backends supported by the core library for a given class. User can pass either one of these values as
backend=...torun()method, or a custom object that hasapplymethod (see documentation forrun()):‘serial’: no parallelization
‘multiprocessing’: parallelization using multiprocessing.Pool
‘dask’: parallelization using dask.delayed.compute(). Requires installation of mdanalysis[dask]
If you want to add your own backend to an existing class, pass a
backends.BackendBasesubclass (see its documentation to learn how to implement it properly), and specifyunsupported_backend=True.- Returns:
names of built-in backends that can be used in
run(backend=...)()- Return type:
Added in version 2.8.0.
- property parallelizable
Boolean mark showing that a given class can be parallelizable with split-apply-combine procedure. Namely, if we can safely distribute
_single_frame()to multiple workers and then combine them with a proper_conclude()call. If set toFalse, no backends except forserialare supported.Note
If you want to check parallelizability of the whole class, without explicitly creating an instance of the class, see
_analysis_algorithm_is_parallelizable. Note that you setting it to other value will break things if the algorithm behind the analysis is not trivially parallelizable.- Returns:
if a given
AnalysisBasesubclass instance is parallelizable with split-apply-combine, or not- Return type:
Added in version 2.8.0.
- run(start: int = None, stop: int = None, step: int = None, frames: Iterable = None, verbose: bool = None, n_workers: int = None, n_parts: int = None, backend: str | BackendBase = None, *, unsupported_backend: bool = False, progressbar_kwargs=None)
Perform the calculation
- Parameters:
start (int, optional) – start frame of analysis
stop (int, optional) – stop frame of analysis
step (int, optional) – number of frames to skip between each analysed frame
frames (array_like, optional) –
array of integers or booleans to slice trajectory;
framescan only be used instead ofstart,stop, andstep. Setting bothframesand at least one ofstart,stop,stepto a non-default value will raise aValueError.Added in version 2.2.0.
verbose (bool, optional) – Turn on verbosity
progressbar_kwargs (dict, optional) – ProgressBar keywords with custom parameters regarding progress bar position, etc; see
MDAnalysis.lib.log.ProgressBarfor full list. Available only forbackend='serial'backend (Union[str, BackendBase], optional) –
By default, performs calculations in a serial fashion. Otherwise, user can choose a backend:
stris matched to a builtin backend (one ofserial,multiprocessinganddask), or aMDAnalysis.analysis.results.BackendBasesubclass.Added in version 2.8.0.
n_workers (int) –
positive integer with number of workers (processes, in case of built-in backends) to split the work between
Added in version 2.8.0.
n_parts (int, optional) –
number of parts to split computations across. Can be more than number of workers.
Added in version 2.8.0.
unsupported_backend (bool, optional) –
if you want to run your custom backend on a parallelizable class that has not been tested by developers, by default False
Added in version 2.8.0.
Changed in version 2.2.0: Added ability to analyze arbitrary frames by passing a list of frame indices in the frames keyword argument.
Changed in version 2.5.0: Add progressbar_kwargs parameter, allowing to modify description, position etc of tqdm progressbars
Changed in version 2.8.0: Introduced
backend,n_workers,n_partsandunsupported_backendkeywords, and refactored the method logic to support parallelizable execution.
- class MDAnalysis.analysis.rms.RMSF(atomgroup, **kwargs)[source]
Calculate RMSF of given atoms across a trajectory.
Note
No RMSD-superposition is performed; it is assumed that the user is providing a trajectory where the protein of interest has been structurally aligned to a reference structure (see the Examples section below). The protein also has be whole because periodic boundaries are not taken into account.
Run the analysis with
RMSF.run(), which stores the results in the arrayRMSF.results.rmsf.- Parameters:
- Raises:
ValueError – raised if negative values are calculated, which indicates that a numerical overflow or underflow occured
Notes
The root mean square fluctuation of an atom \(i\) is computed as the time average
\[\rho_i = \sqrt{\left\langle (\mathbf{x}_i - \langle\mathbf{x}_i\rangle)^2 \right\rangle}\]No mass weighting is performed.
This method implements an algorithm for computing sums of squares while avoiding overflows and underflows [1].
Examples
In this example we calculate the residue RMSF fluctuations by analyzing the \(\text{C}_\alpha\) atoms. First we need to fit the trajectory to the average structure as a reference. That requires calculating the average structure first. Because we need to analyze and manipulate the same trajectory multiple times, we are going to load it into memory using the
MemoryReader. (If your trajectory does not fit into memory, you will need to write out intermediate trajectories to disk or generate an in-memory universe that only contains, say, the protein):import MDAnalysis as mda from MDAnalysis.analysis import align from MDAnalysis.tests.datafiles import TPR, XTC u = mda.Universe(TPR, XTC, in_memory=True) protein = u.select_atoms("protein") # 1) the current trajectory contains a protein split across # periodic boundaries, so we first make the protein whole and # center it in the box using on-the-fly transformations import MDAnalysis.transformations as trans not_protein = u.select_atoms('not protein') transforms = [trans.unwrap(protein), trans.center_in_box(protein, wrap=True), trans.wrap(not_protein)] u.trajectory.add_transformations(*transforms) # 2) fit to the initial frame to get a better average structure # (the trajectory is changed in memory) prealigner = align.AlignTraj(u, u, select="protein and name CA", in_memory=True).run() # 3) reference = average structure ref_coordinates = u.trajectory.timeseries(asel=protein).mean(axis=1) # make a reference structure (need to reshape into a 1-frame # "trajectory") reference = mda.Merge(protein).load_new(ref_coordinates[:, None, :], order="afc")
We created a new universe
referencethat contains a single frame with the averaged coordinates of the protein. Now we need to fit the whole trajectory to the reference by minimizing the RMSD. We useMDAnalysis.analysis.align.AlignTraj:aligner = align.AlignTraj(u, reference, select="protein and name CA", in_memory=True).run()
The trajectory is now fitted to the reference (the RMSD is stored as aligner.results.rmsd for further inspection). Now we can calculate the RMSF:
from MDAnalysis.analysis.rms import RMSF calphas = protein.select_atoms("name CA") rmsfer = RMSF(calphas, verbose=True).run()
and plot:
import matplotlib.pyplot as plt plt.plot(calphas.resnums, rmsfer.results.rmsf)
References
Added in version 0.11.0.
Changed in version 0.16.0: refactored to fit with AnalysisBase API
Deprecated since version 0.16.0: the keyword argument quiet is deprecated in favor of verbose.
Changed in version 0.17.0: removed unused keyword weights
Changed in version 1.0.0: Support for the
start,stop, andstepkeywords has been removed. These should instead be passed toRMSF.run().- results.rmsf
Results are stored in this N-length
numpy.ndarrayarray, giving RMSFs for each of the given atoms.Added in version 2.0.0.
- rmsf
Alias to the
results.rmsfattribute.Deprecated since version 2.0.0: Will be removed in MDAnalysis 3.0.0. Please use
results.rmsfinstead.
- classmethod get_supported_backends()[source]
Tuple with backends supported by the core library for a given class. User can pass either one of these values as
backend=...torun()method, or a custom object that hasapplymethod (see documentation forrun()):‘serial’: no parallelization
‘multiprocessing’: parallelization using multiprocessing.Pool
‘dask’: parallelization using dask.delayed.compute(). Requires installation of mdanalysis[dask]
If you want to add your own backend to an existing class, pass a
backends.BackendBasesubclass (see its documentation to learn how to implement it properly), and specifyunsupported_backend=True.- Returns:
names of built-in backends that can be used in
run(backend=...)()- Return type:
Added in version 2.8.0.
- property parallelizable
Boolean mark showing that a given class can be parallelizable with split-apply-combine procedure. Namely, if we can safely distribute
_single_frame()to multiple workers and then combine them with a proper_conclude()call. If set toFalse, no backends except forserialare supported.Note
If you want to check parallelizability of the whole class, without explicitly creating an instance of the class, see
_analysis_algorithm_is_parallelizable. Note that you setting it to other value will break things if the algorithm behind the analysis is not trivially parallelizable.- Returns:
if a given
AnalysisBasesubclass instance is parallelizable with split-apply-combine, or not- Return type:
Added in version 2.8.0.
- run(start: int = None, stop: int = None, step: int = None, frames: Iterable = None, verbose: bool = None, n_workers: int = None, n_parts: int = None, backend: str | BackendBase = None, *, unsupported_backend: bool = False, progressbar_kwargs=None)
Perform the calculation
- Parameters:
start (int, optional) – start frame of analysis
stop (int, optional) – stop frame of analysis
step (int, optional) – number of frames to skip between each analysed frame
frames (array_like, optional) –
array of integers or booleans to slice trajectory;
framescan only be used instead ofstart,stop, andstep. Setting bothframesand at least one ofstart,stop,stepto a non-default value will raise aValueError.Added in version 2.2.0.
verbose (bool, optional) – Turn on verbosity
progressbar_kwargs (dict, optional) – ProgressBar keywords with custom parameters regarding progress bar position, etc; see
MDAnalysis.lib.log.ProgressBarfor full list. Available only forbackend='serial'backend (Union[str, BackendBase], optional) –
By default, performs calculations in a serial fashion. Otherwise, user can choose a backend:
stris matched to a builtin backend (one ofserial,multiprocessinganddask), or aMDAnalysis.analysis.results.BackendBasesubclass.Added in version 2.8.0.
n_workers (int) –
positive integer with number of workers (processes, in case of built-in backends) to split the work between
Added in version 2.8.0.
n_parts (int, optional) –
number of parts to split computations across. Can be more than number of workers.
Added in version 2.8.0.
unsupported_backend (bool, optional) –
if you want to run your custom backend on a parallelizable class that has not been tested by developers, by default False
Added in version 2.8.0.
Changed in version 2.2.0: Added ability to analyze arbitrary frames by passing a list of frame indices in the frames keyword argument.
Changed in version 2.5.0: Add progressbar_kwargs parameter, allowing to modify description, position etc of tqdm progressbars
Changed in version 2.8.0: Introduced
backend,n_workers,n_partsandunsupported_backendkeywords, and refactored the method logic to support parallelizable execution.